RNA-seq数据分析流程
普通RNA-seq数据分析
RNA-seq数据分析流程
(一)原始数据的质控
以其中一个样本为例(其他样本指控结果基本类似)
1 | cd /home/raw-data |
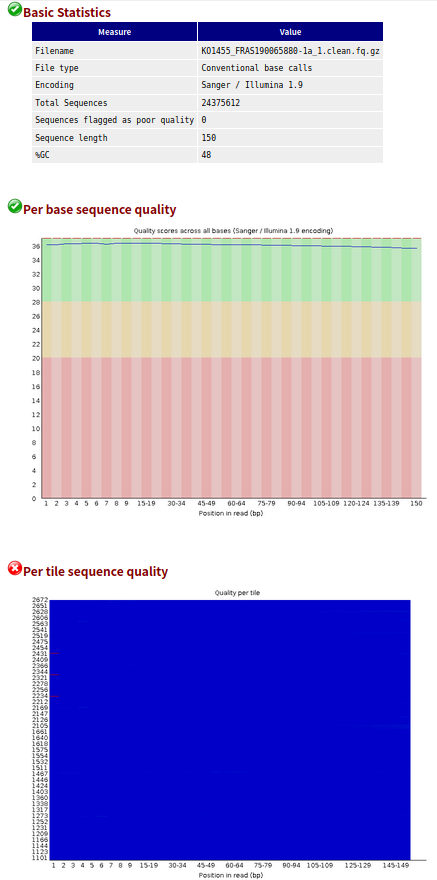
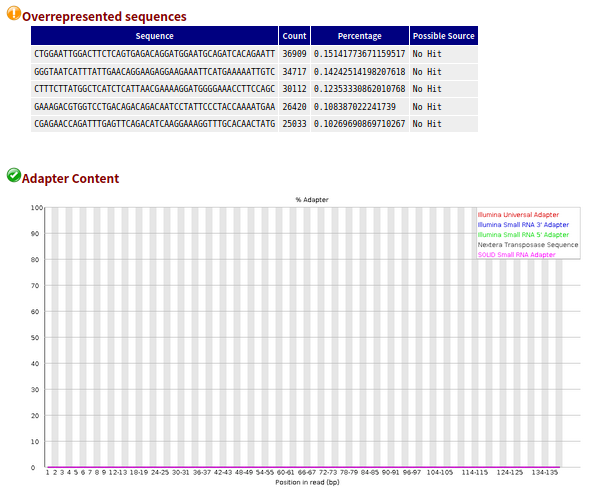
- 1、帮忙分析的数据,从公司给的结果的样本名中包含“clean”,我猜想公司大概已经进行过一次过滤,而且也完全没有接头。
- 2、另外,没有过关的这些数据也很难通过过滤得到改善(因为我使用trim_galore过滤,质控结果更差了)
- 3、但是我还是不死心呀,我分别取了一个原始的和自己过滤的样本的前10000条reads,进行比对。结果发现原始的比对率可以达到95%+,自己过滤后的样本比对率比较低。
- 4、所以,这批数据,公司应该已经进行过滤操作了。
(二)比对
比对软件:hisat2
不同数据类型所需要使用的比对软件也是有讲究的呢,可千万不能乱使用,会出问题的,刚开始接触数据分析时,知道有很多比对软件,但一直用bowtie2进行比对,后面就翻车啦~~~~~~
比对软件的选择:
https://www.jianshu.com/p/849f8ada0ab7
https://www.jianshu.com/p/681e02e7f9af
https://zhuanlan.zhihu.com/p/26506787
NGS—-bwa
Chip(也包括ATAC)—-bowtie2
RNA—-hisat2(推荐)、star、Tophat、subjunc
第一步:序列比对
1 | #cat sample |
1 | #因为不想让本机太辛苦,要让它歇息一会儿,所以将KO和WT分批次跑 |
第二步:构建索引
1 | ls *.bam|xargs -i samtools index {} |
第三部:生成状态文件(统计比对情况)
1 | ls *.bam|xargs -i samtools flagstat -@ 7 {} |
(三)制作表达矩阵
参考资料:https://www.bioinfo-scrounger.com/archives/407/
1 | ref=/home/reference/gtf/gencode.v33.annotation.gtf |
查看featureCounts的情况
1 | cat counts.id.log |
在此,要记录一个错误。第一次操作时误使用了人类的gtf文件,结果日志文件显示比对率在10%左右……使用小鼠gtf文件后,比对结果正常多了(因为这批数据是小鼠组织细胞测序的结果)。如下所示。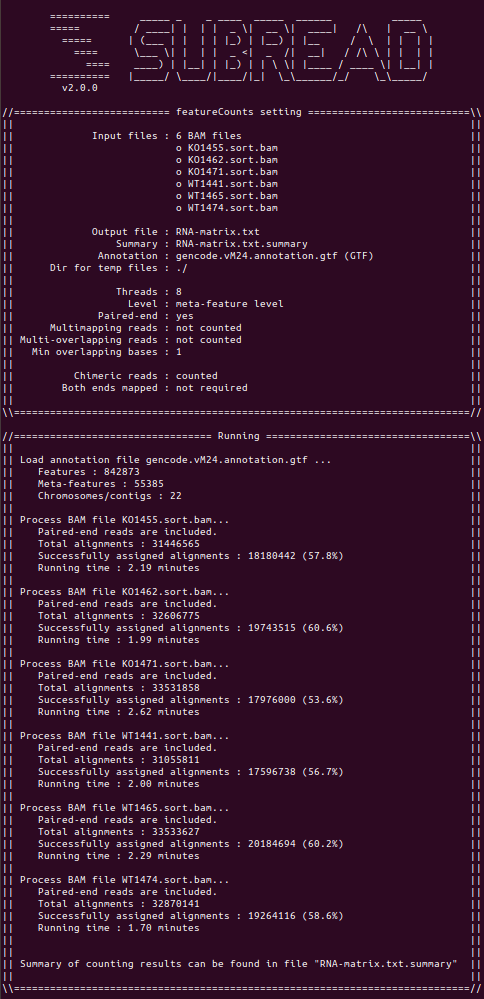
(四)差异分析
第一步:将reads矩阵导入R中
1 | rm(list = ls()) |
第二步:获取正确的Geneid
- 首先看看矩阵中的Geneid长什么样
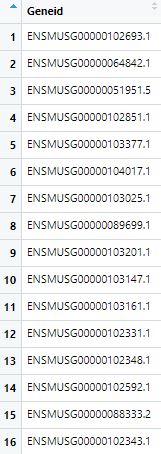
- 所以要去掉小数点及后面的数字,获取正确的Geneid表达矩阵现在的样子(Geneid名去掉了多余的部分)
1
2
3
4
5
6
7library(stringr)
class(str_split(a$Geneid,'[.]',simplify = T))
a$ensembl_id=str_split(a$Geneid,'[.]',simplify = T)[,1]
rownames(a)<-a$ensembl_id
check_a_again=a[1:4,1:12]
exprSet<-a[,7:12]
ckeck_exprSet<-head(exprSet)
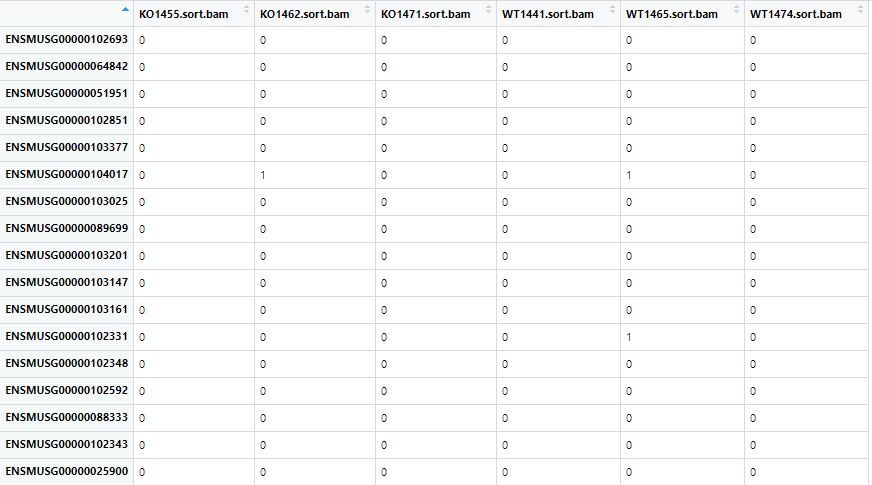
第三步:id转换
Geneid和symbol并不是完全一一对应的,存在部分多个Geneid对应同一个基因名的情况。
因为rownames必须唯一,所以就要去掉重复的Geneid或者只保留其中一个(这个保留就比较的人为了~)
方法一:使用biomaRt包
此方法几乎可以转换全部的id(推荐)
1 | library(biomaRt) |
- 方法二:使用org.Mm.eg.db包
可能因为此包不怎么更新,id转换率只有50%左右,这样回丢掉很多信息的,可能会导致后续分析不可靠(不推荐)
1 | library(clusterProfiler) |
https://www.jianshu.com/p/3a0e1e3e41d0
https://www.jianshu.com/p/c06fea33b60f
- 在进行后续分析之前,检验目的基因的敲除效果
1
2
3
4
5
6
7
8
9
10
11
12
13
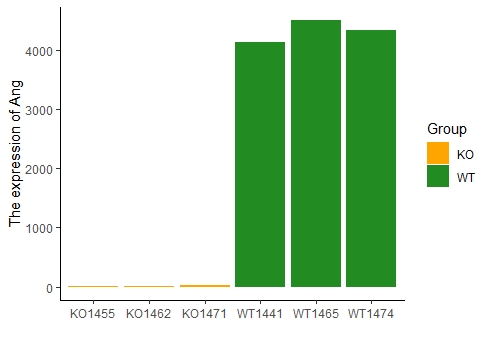
14#Ang表达量探索
Ang<-raw_exprSet[rownames(raw_exprSet)=="Ang",]
colnames(Ang)<-c("KO1455","KO1462","KO1471","WT1441","WT1465","WT1474")
ang<-as.data.frame(t(Ang))
ang$Group<-c(rep("KO",3),rep("WT",3))
ang$Sample<-rownames(ang)
colnames(ang)<-c("Counts","Group","Sample")
library(ggplot2)
p<-ggplot(ang,aes(Sample,Counts,group=Group))+
geom_bar(aes(fill=Group),stat='identity')+
scale_fill_manual(values = c("#FFA500","#228B22"))+
ylab("The expression of Ang")+
xlab("")
p+theme_bw()+theme(panel.border = element_blank(),panel.grid.major = element_blank(),panel.grid.minor = element_blank(),axis.line = element_line(colour = "black"))
由图可看出:目的基因敲除效果显著;另外,也可以一定程度上反应上游数据分析的可靠性(因为在做测序之前,一定是先实验验证过敲低效率的)。
https://zhuanlan.zhihu.com/p/92473504
https://www.sohu.com/a/319083452_785442
第四步:DESeq2进行差异分析
1、DESeq2差异分析的主要步骤
1 | library(DESeq2) |
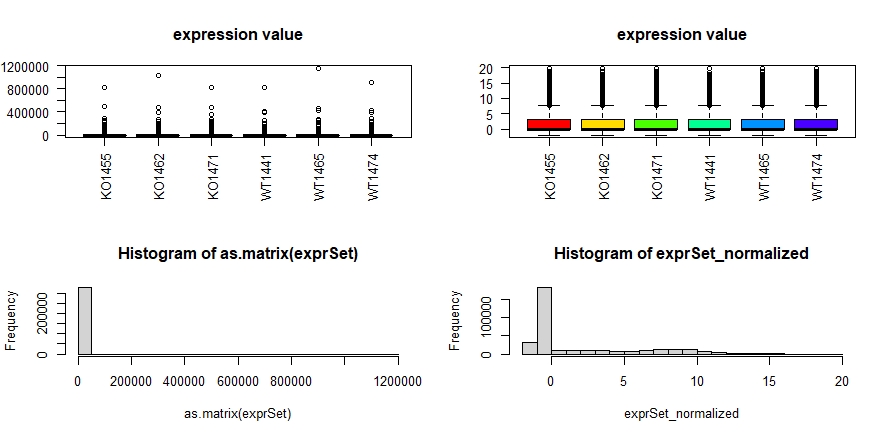
2、DESeq2标准化前后的对比
1 | #数据离散度非常大的RNA-seq的基因的reads的counts矩阵经过normalization后 |
https://zhuanlan.zhihu.com/p/30350531
第五步:可视化 ……
- 富集分析(可以使用KEGG、GO,这里使用GO)
大致看一下受目的基因影响的信号通路1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28#1、差异基因的提取
topT <- as.data.frame(DEG)
logFC_Cutoff<-with(topT,mean(abs(log2FoldChange))+2*sd(abs(log2FoldChange)))
logFC_cutoff<-logFC_Cutoff
select_gene=(abs(DEG$log2FoldChange)>=logFC_cutoff)&(DEG$pvalue<0.05)
select_gene.sig=DEG[select_gene,]
write.csv(select_gene.sig,'DESeq2_select_gene.sig.csv')
gene=rownames(select_gene.sig)
#2、差异基因的通路富集
library(clusterProfiler)
library(topGO)
library(Rgraphviz)
library(pathview)
library(org.Mm.eg.db)
DEG.entrez_id = mapIds(x = org.Mm.eg.db,
keys = gene,
keytype = "SYMBOL",
column = "ENTREZID")
DEG.entrez_id = na.omit(DEG.entrez_id)
erich.go.BP = enrichGO(gene = DEG.entrez_id,
OrgDb = org.Mm.eg.db,
keyType = "ENTREZID",
ont = "BP",
pvalueCutoff = 0.5,
qvalueCutoff = 0.5)
##分析完成后,作图
dotplot(erich.go.BP)
https://www.jianshu.com/p/47b5ea646932?utm_source=desktop&utm_medium=timeline
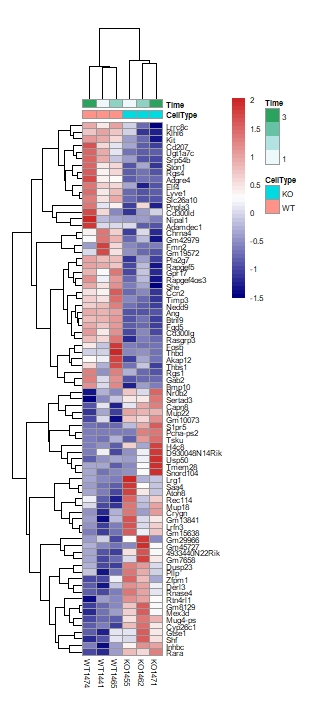
- 热图绘制
1
2
3
4
5
6
7
8
9
10
11library(pheatmap)
colnames(exprSet)<-c("KO1455","KO1462","KO1471","WT1441","WT1465","WT1474")
choose_upgene=names(sort(apply(up_gene, 1, mad),decreasing = T)[1:40])
choose_upgene_matrix=exprSet[choose_upgene,]
choose_downgene=names(sort(apply(down_gene, 1, mad),decreasing = T)[1:40])
choose_downgene_matrix=exprSet[choose_downgene,]
choose_matrix=rbind(choose_upgene_matrix,choose_downgene_matrix)
choose_matrix=t(scale(t(choose_matrix)))
annotation_col = data.frame(CellType = factor(rep(c("KO", "WT"), each=3)),Time = 1:3)
rownames(annotation_col)<-colnames(exprSet)
pheatmap(choose_matrix,fontsize = 6,cexCol=1,cellwidth=10,cellheigh=5,color = colorRampPalette(c('navy','white','firebrick3'))(50),annotation_col=annotation_col)
RNA-seq数据分析流程